Dimer Method
Overview
The Dimer method is a minimum-mode following algorithm for locating first-order saddle points on the potential energy surface. Unlike path-based methods such as NEB or String, the Dimer method requires only a single starting structure and does not need knowledge of the product geometry.
The method works by rotating a trial direction toward the lowest curvature mode of the PES, then translating the structure along that mode. The current runtime uses calculator Hessian-vector products through get_hvp() for this rotation/translation loop, so Dimer requires a model backend that exposes that capability. Among shipped MAPLE backends, this currently means ANI in gas phase.
Parameters
| Parameter | Type | Default | Description |
|---|---|---|---|
use_hvp |
bool | False | Compatibility flag. The current Dimer execution path requires calculator get_hvp() support regardless of this setting; shipped support is ANI gas phase only. |
delta |
float | 0.005 | Legacy finite-difference displacement. It is not used by the current get_hvp()-based execution path. |
rot_max_iter |
int | 5 | Maximum rotation iterations per translation step. |
rot_alpha |
float | 0.5 | Rotation step scaling factor. |
rot_f_max_th |
float | 1.0e-3 | Max force threshold for rotation convergence. |
rot_f_rms_th |
float | 5.0e-4 | RMS force threshold for rotation convergence. |
step0 |
float | 0.2 | Initial translation step size. |
step_max |
float | 0.15 | Maximum translation step size. |
trust_radius |
float | 0.15 | Trust radius for translation. |
f_max_th |
float | 5.0e-3 | Max force convergence threshold for overall dimer. |
f_rms_th |
float | 1.0e-3 | RMS force convergence threshold. |
kappa_to_flip |
float | 0.0 | Curvature threshold for mode flipping. |
max_iter |
int | 200 | Maximum dimer translation iterations. |
use_mass_weight |
bool | False | Use mass-weighted coordinates. |
remove_rigid |
bool | True | Remove rigid body translations and rotations. |
n_init |
str | "random" | Initial dimer direction: "random", "force", or "given". |
n_given |
ndarray | None | User-supplied initial dimer direction vector. |
save_traj |
bool | True | Save trajectory of dimer steps. |
save_metrics |
bool | True | Save convergence metrics to output. |
Input Example
Checked-in MAPLE example: examples/ts/dimer/inp1.inp. Dimer is single-ended, so the input is one starting geometry rather than reactant/product endpoints.
#model=ANI-1xnr
#ts(method=dimer)
#device=gpu0
C 0.8773307780 0.7831579390 -0.1629806432
H 0.6909163982 1.5540583811 -0.9219751886
O 0.5296065367 0.9762287325 1.1234790578
C 1.3488671845 -0.5563869274 -0.1923254310
H 0.7828987178 -0.2429788233 1.0663324170
H 1.0764966981 -1.2943318365 -0.9583627957
H 2.2951253096 -0.7293808119 0.3397718379Dimer is single-ended, but it is not model-agnostic: the active calculator must implement get_hvp(). If a selected model does not provide Hessian-vector products, choose P-RFO/NEB/String instead or switch to ANI gas phase.
Output
*_dimer_ts.xyz— The final converged transition state geometry.*_dimer_traj.xyz— Multi-frame XYZ trajectory file containing all intermediate structures generated during the search from the initial guess to the saddle point.
Visualization
The Dimer trajectory visualization uses *_dimer_traj.xyz, while the final TS panel uses *_dimer_ts.xyz. Because Dimer is single-ended, the trajectory shows the search process rather than a reactant-to-product MEP.
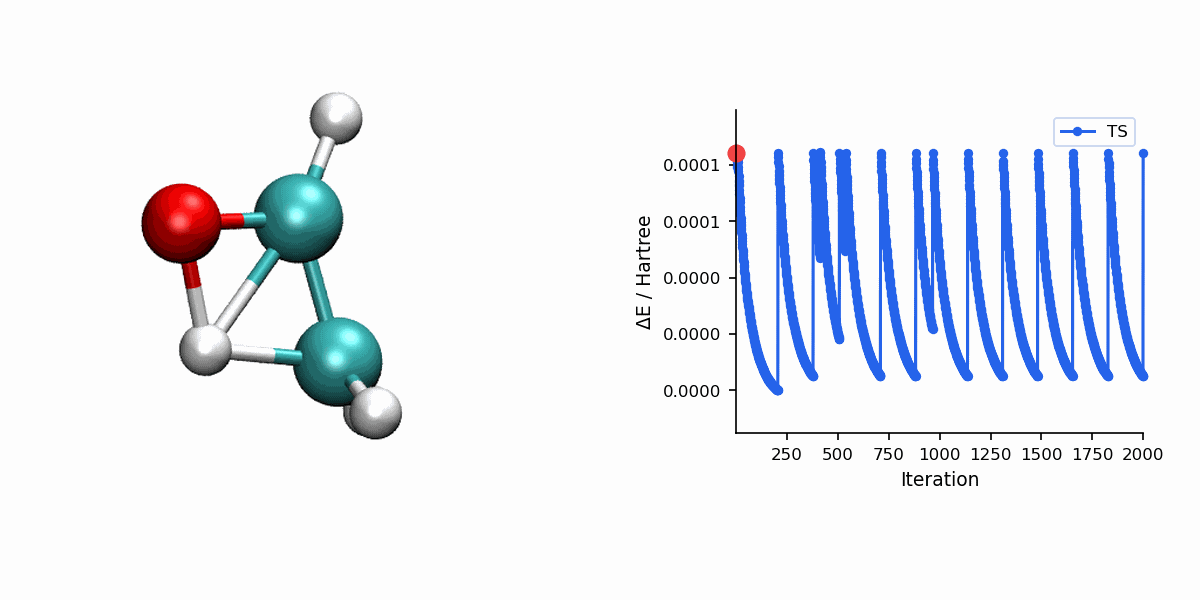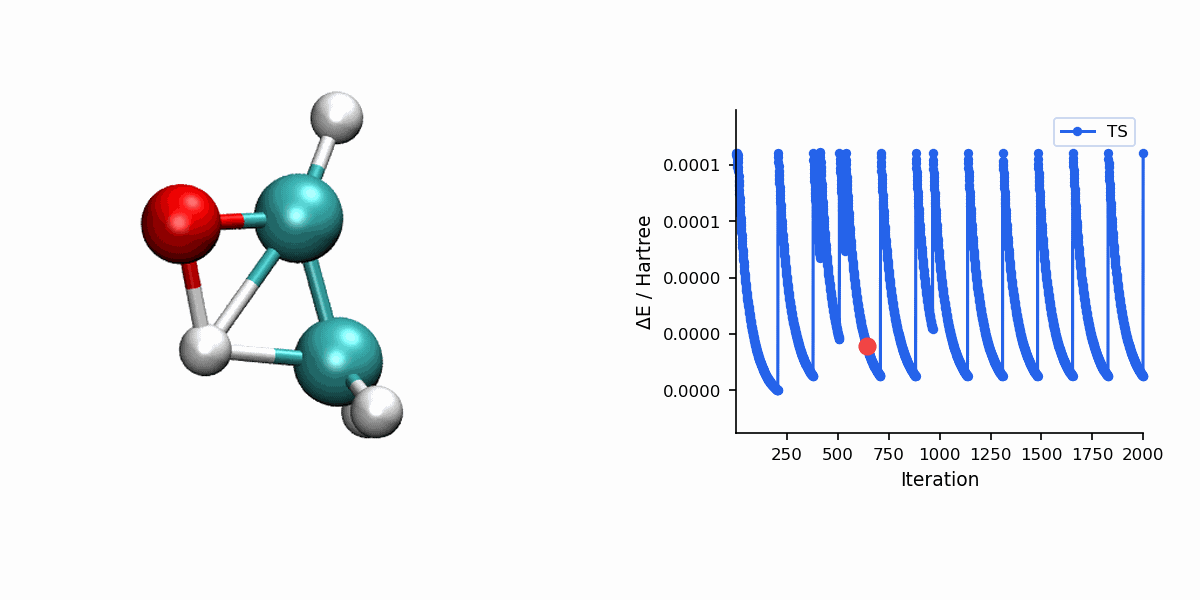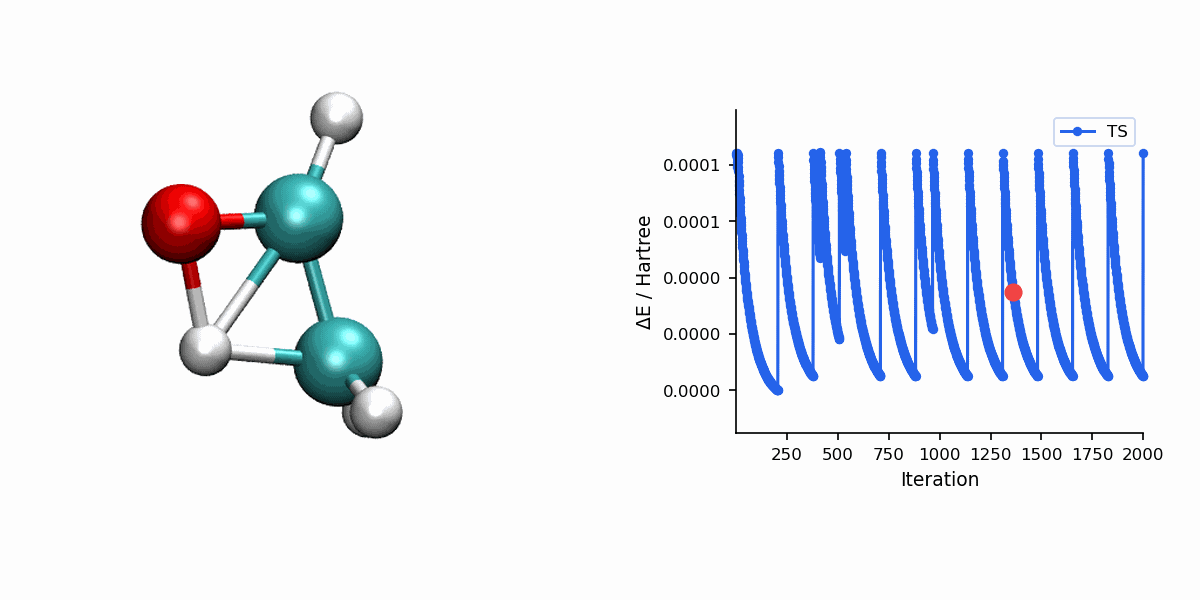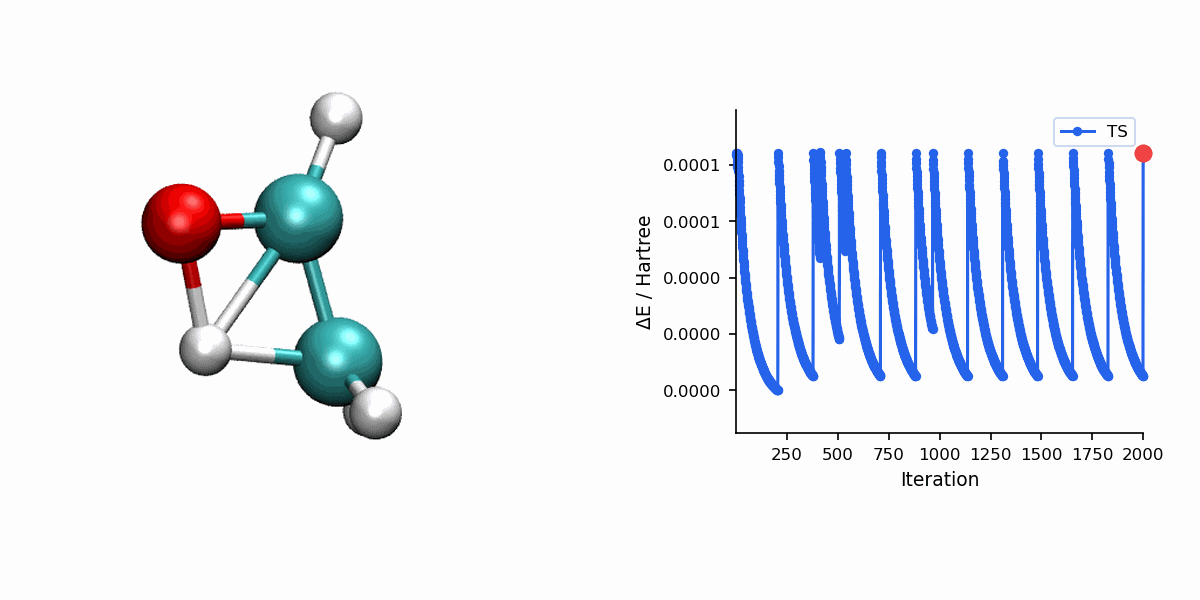
When to Use
The Dimer method is the right choice when:
- The product is unknown. You want to explore what reactions are possible from a given structure without knowing the endpoint geometry in advance.
- Exploring the PES landscape. You want to systematically discover nearby saddle points from a minimum, for example to enumerate possible decomposition pathways or rearrangement channels.
- Single-ended search is preferred. You have a reasonable guess for the TS geometry (e.g., from chemical intuition or a constrained scan) but do not have a product structure to use with NEB or String.
- The reaction path is not well-defined. When the reaction involves complex conformational changes where defining a clear product is difficult, the Dimer method can find the nearest saddle point without requiring path construction.