MAPLE
Machine-learning Potential for Landscape Exploration
A fast, GPU-accelerated computational chemistry toolkit that combines state-of-the-art machine learning potentials with practical workflows for structures, energies, reaction paths, and molecular landscapes.
What is MAPLE?
MAPLE is a next-generation computational chemistry toolkit designed to make machine learning potentials practical and powerful for real-world research. By uniting accurate ML models, GPU acceleration, and an integrated workflow, MAPLE enables rapid and reliable exploration of molecular systems—from small molecules to complex landscapes.
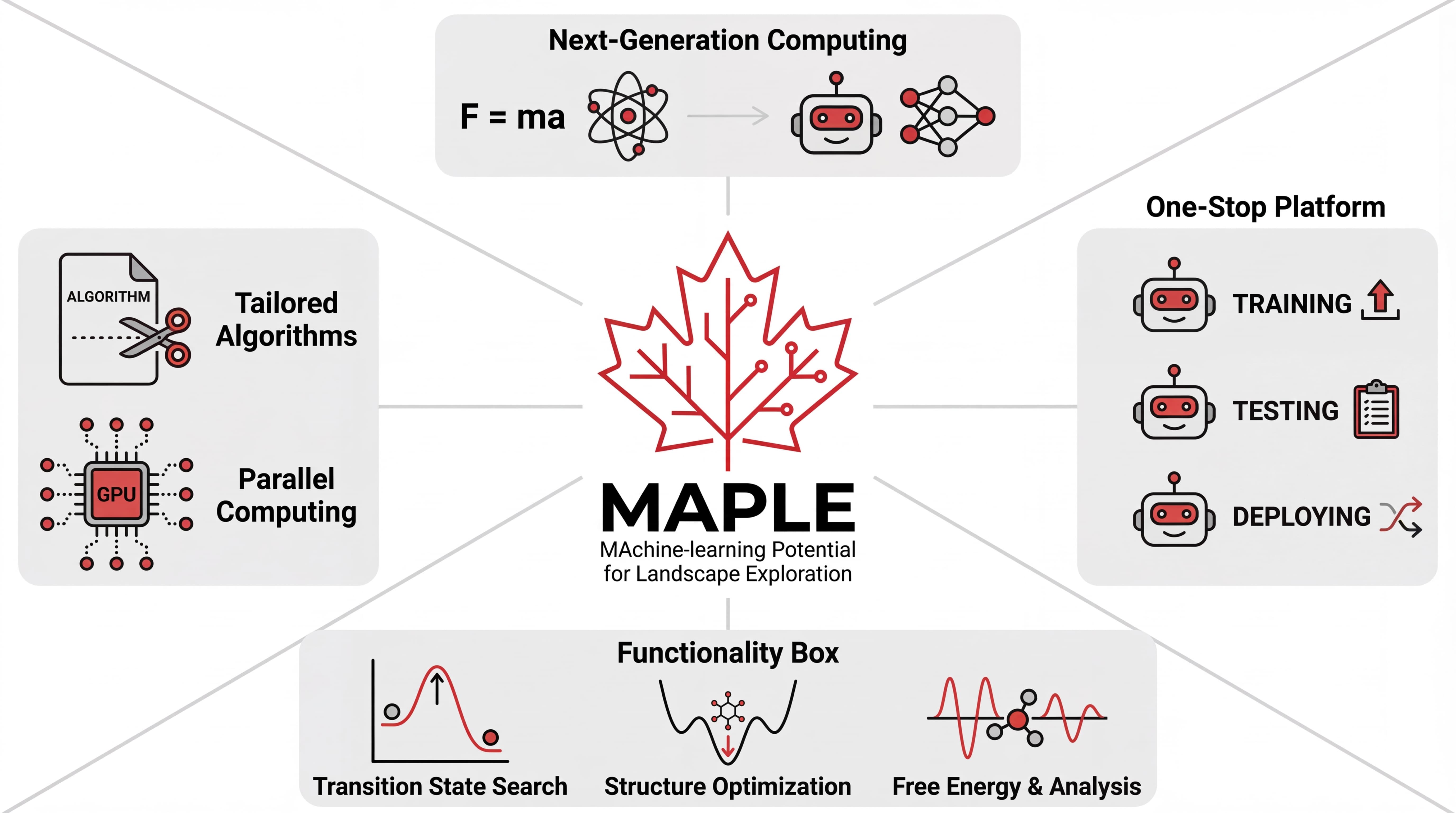
Built for practical molecular landscape exploration
MAPLE brings the original toolkit highlights back to the front: models, optimization, transition states, reaction paths, spectra, scans, solvation, and GPU acceleration.
ML Potentials
Support for ANI, AIMNet2, MACE-OFF, UMA, and more, with backend-specific capabilities documented in the calculator matrix.
Geometry Optimization
L-BFGS, RFO, steepest descent, conjugate gradient, DIIS, and bracketing L-BFGS methods.
Transition States
NEB, CI-NEB, P-RFO, Dimer, Growing String, AutoNEB methods for saddle point search.
Reaction Paths
IRC with GS, LQA, HPC, and EulerPC integrators. Verify TS connectivity to reactants and products.
Vibrational Analysis
Normal mode analysis, RRHO thermochemistry, IR spectra. Mass-weighted Hessian diagonalization.
PES Scanning
1D, 2D, or 3D relaxed or rigid scans. Map energy landscapes along chosen coordinates.
Solvation
Experimental implicit single-point solvent energies and non-periodic explicit solvent clusters.
GPU Accelerated
CUDA support for ML potential evaluation. Orders of magnitude faster than traditional QM.
More Coming Soon
Additional features are actively under development. Stay tuned for future releases.
Supported ML Potentials
MAPLE integrates leading machine learning potentials to cover a broad range of systems.
Computational Capabilities
Powerful tools for exploring structures, energies, and reaction pathways.
Additional Features
Advanced options that enhance control, accuracy, and performance.
Articles & News
Recent publications and release milestones from the MAPLE project.
An MLFF-native platform for automated reaction modeling and enzyme design.
April 1, 2026 MAPLE v0.1.3 releasedExpanded MD workflows, UMA/MACEPol updates, and IRC/debug cleanup.
February 7, 2026 MAPLE v0.1.1 releasedAutoNEB, NVE MD, SDCG, and new IRC integrators.
December 31, 2025 First public releaseCore optimization, transition-state, IRC, and frequency tools.